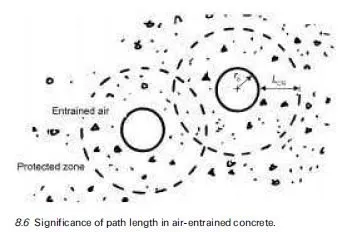
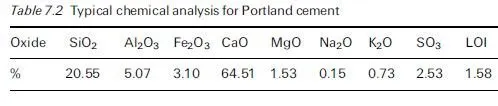
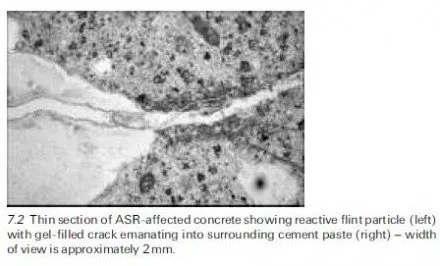
Introduction
The chemistry of the pore solution resident in the pores of a given concrete may strongly influence its potential durability. The potential for development of ASR, steel corrosion, sulfate attack, `sea water attack’ and concrete spalling, among others, all directly reflect the influence of pore solution chemistry and of changes that may take place within it. Unfortunately, concrete pore solutions and their potential changes under different field exposure conditions are not particularly well understood by many who are concerned with concrete durability. In this section the writer hopes to provide a realistic treatment of the origin and evolution of pore solutions in concretes, and of some of the changes that may be induced in them by exposures to different external environments.
Genesis and early development of pore solutions
When concrete is mixed, chemical reactions occur immediately after the mix water is added. The water used is typically specified as `potable’ water; these reactions quickly result in a drastic change in its chemical character. Within a very few minutes this `potable water’ becomes a high ionic strength solution of greatly altered chemical properties. The chemistry of this solution depends very much on the chemistry of the particular cement used. Before setting takes place, this concentrated `mix solution’ constitutes the continuous phase in which aggregate and cement particles are suspended. On setting, the mix solution is seamlessly carried over to become the `pore solution’ contained within the concrete pores. The composition of this mix solution/pore solution evolves over time, with major changes occurring especially during the first day. This evolution can be readily followed in the laboratory. It is possible to separate mix solution for chemical analysis from freshly-mixed cement paste (or concrete) by gas pressure-assisted filtration. Repeated sampling can be carried out at intervals until the approach of setting renders it impractical. After setting and some strength gain, pore solutions can again be expressed for analysis, for example by the pore solution expression equipment described by Barneyback and Diamond.2 Table 2.1 provides an illustrative set of the results of analyses of such a series of mix/pore solutions developed in a Portland cement paste that might be found in an archetypical concrete. The paste was prepared from a relatively low alkali Portland cement (0.45% Na2Oequiv.), and was mixed at a w:c ratio of 0.5. The data were taken from the thesis of Penko.3 The ionic species listed in Table 2.1 are universally found in significant concentrations in the mix/pore solutions developed during the early hydration of Portland cements. These are K+, Na+, Ca2+, SO4 2±, and OHÿ. Aluminum, iron, and silicate ions are present only in concentrations that are orders of magnitude lower than those of the five listed.
The high concentrations of alkali metal ions and of sulfate ions seen in Table 2.1 are derived from the alkali sulfate `impurities’ carried by the cement. Even with the comparatively low alkali content of the particular cement, the early sulfate ion concentration found in solution vastly exceeds that of a saturated solution of gypsum. More importantly, it also exceeds that of ettringite; and ettringite will precipitate rapidly as aluminate ions become available following the dormant period. For the paste of Table 2.1, the dormant period ended at a little less than three hours after mixing. Initial set occurred at ca. 3.5 hours, and final set at slightly over 5 hours. The peak temperature indicative of the maximum rate of hydration was measured at 8.5 hours. It is seen that almost equal concentrations of hydroxide and sulfate ions were present throughout the early stage of hydration. Nevertheless, the presence of the hydroxide ions from the time of earliest analysis makes it evident that earliest reaction was not confined to dissolution of alkali sulfates, but included reaction with C3S as well. With respect to the alkali metal cations, it is seen that the K+ concentration vastly exceeds the Na+ concentration at all stages. This is a feature common in modern cements, but not universally so. It is seen in Table 2.1 that little change takes place in concentrations of any of the ions during at least the first six hours, despite the fact that within this period the dormant period ended and both initial set and final set occurred. This is a common pattern; neither the onset of active hydration after the dormant period nor the occurrence of setting produces significant changes in mix/pore solution ion concentrations. Some years ago it was established that rapid ettringite formation after the end of the dormant period quickly removes sulfate from the pore solution, but that the sulfate lost from solution is continually replaced by progressive dissolution of the gypsum.4 Thus the dissolved sulfate ion concentration is maintained at least approximately constant as long as some solid gypsum persists, apparently in response to an equilibrium involving the simultaneous presence of syngenite, gypsum and ettringite. The dissolution of the last of the solid gypsum marks a turning point; subsequent ettringite precipitation progressively reduces the sulfate concentration of the pore solution ± in properly formulated Portland cements ± to very low levels by the end of the first day. For the specific paste of Table 2.1, the turning point marked by the depletion of the solid gypsum apparently occurred some time shortly after 6 hours. A crucial feature of the post-turning point changes in pore solution composition is that, as the sulfate concentration is depleted, electrical neutrality is maintained by parallel increases in the OHÿ ion concentration, rather than by reductions in the alkali cation concentrations. Thus the pH goes up sharply as the sulfate is depleted from solution. As the pH goes up, the already relatively modest calcium ion concentration (modest in absolute terms, not in degree of supersaturation) is reduced to a very low value. The result of these changes is that the pre-existing mix/pore solution is progressively transformed into a concentrated solution of potassium and sodium hydroxide. This transformation has highly significant consequences with respect to possible development of ASR and with respect to the maintenance of steel passivation in concrete, among other effects. It appears that this enhancement of pH, while universal in properly for- mulated Portland cements, is not inevitable. It has been shown3 that if sufficient excess gypsum is added to the cement, the exhaustion of the available aluminate can end the precipitation of ettringite before the solid gypsum is fully depleted. Under these circumstances the usual pH transformation can be postponed or perhaps prevented indefinitely. Conversely, in the absence of gypsum, as, for example, in laboratory experiments when ground clinker is hydrated without any inter-ground gypsum, the decline of sulfate ion concentration and the concomitant pH increase begin almost immediately after mixing, and are quickly completed.